Elastic network model of proteins with python
I used this two documents to write this post:
- Low-frequency normal modes of proteins. Yves-Henri Sanejouand. Biological Physics. Université Claude Bernard - Lyon I, 2007. In french.
- Normal mode theory and harmonic potential approximations. Konrad Hinsen
Other documents of interest:
A harmonic potential well has the form:
with a dimensional vector ( is the number of atoms) of the stable conformation and the same object of the current conformation.
is the Hessian matrix of :
and:
We denote:
and
Now we can define the second derivative of :
and
which allows us to define the Hessian matrix :
as:
Which can then be written as,
The diagonal of is defined with:
Now we can play with python, a protein, and normal modes. First the import:
import sys
sys.path.append('/home/bougui/SOM')
import IO
import scipy.spatial
from mpl_toolkits.mplot3d import Axes3D
%pylab inlineAnd we load the pdb structure with only atoms.
struct = IO.Structure('data/1E4E-prot-CA.pdb')Now we extract the coordinates of the atoms ():
R = struct.atoms['coord']Then is computed with scipy.spatial and we define a function to
compute the Hessian matrix as defined below. We put a distance cutoff of 15
Angstrom.
def supersum(K,i):
a = zeros((3,3))
N = K.shape[0]/3
for j in range(N):
a+=K[i*3:i*3+3,j*3:j*3+3]
return a
def supertrace(K,i,j):
return trace(K[i*3:i*3+3,j*3:j*3+3])
def hessian(R):
s_squared = scipy.spatial.distance.squareform(scipy.spatial.distance.pdist(R, metric='sqeuclidean'))
nr,nc = s_squared.shape
N = nr # number of atom: nr = nc
K = zeros((N*3,N*3))
for i in range(N):
for j in range(i+1,N):
xyz_i, xyz_j = R[i], R[j]
d_xyz = xyz_j - xyz_i
if s_squared[i,j] < 15**2:
K[i*3:i*3+3,j*3:j*3+3] = -dot(d_xyz[:,None],d_xyz[:,None].T) / s_squared[i,j]
K += K.T
for i in range(N):
a = supersum(K,i)
K[i*3:i*3+3,i*3:i*3+3] = -a
return K
rcParams['figure.figsize'] = 8,8
K = hessian(R)
imshow(K, cmap=cm.PRGn, interpolation='nearest', vmin=-1, vmax=1)
tmp = colorbar()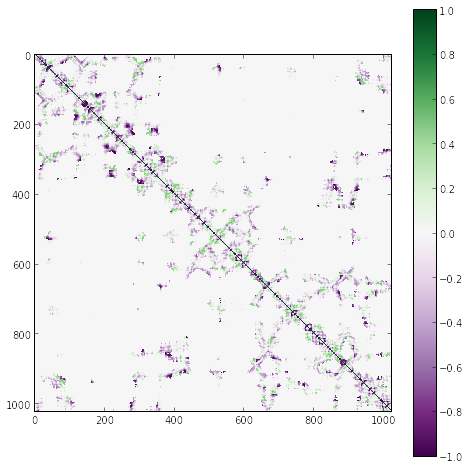
And then we diagonalize the Hessian matrix above:
w,v = eig(K)
v = v[:,w.argsort()]
w = w[w.argsort()]The sorted eigenvalues are plotted:
rcParams['figure.figsize'] = 8,4
plot(w, linewidth=2)
xlabel('eigenvalue index', fontsize=14)
ylabel('$\lambda$', fontsize=18)
grid()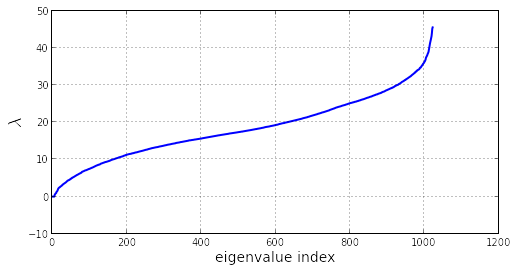
The first 6 eigenvalues are null (the 6 degrees of freedom of a rigid body in a 3D space):
plot(w[:20], linewidth=2)
xlabel('eigenvalue index', fontsize=14)
ylabel('$\lambda$', fontsize=18)
grid()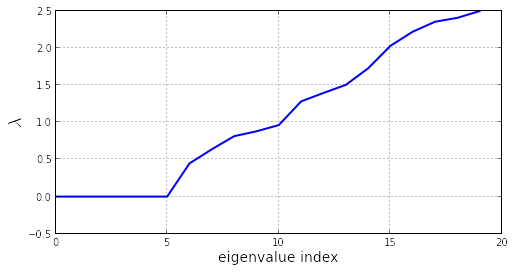
Then, we can compute the covariance and correlation matrix from the inverse of the Hessian matrix :
with the number of considered atoms.
N = K.shape[0]/3
hessian_inv = zeros((3*N,3*N))
for i in range(6,3*N):
hessian_inv += (dot(v[:,i][:,None],v[:,i][:,None].T)/w[i])The element of the covariance matrix is given by the trace of each super element of . The diagonal of the covariance matrix give the amplitude of the fluctuation for each considered atom of the system.
rcParams['figure.figsize'] = 5,10
cov = zeros((N,N))
for i in range(N):
for j in range(N):
cov[i,j] = supertrace(hessian_inv,i,j)
d = cov.diagonal()
subplot(211)
plot(d)
xlabel('sequence of considered atoms', fontsize=14)
ylabel('Amplitude of the fluctuation', fontsize=14)
grid()
corr = cov / sqrt(dot(d[None,:].T,d[None,:]))
subplot(212)
imshow(corr, vmax=0.2)
title('Correlation matrix', fontsize=14)
tmp = colorbar()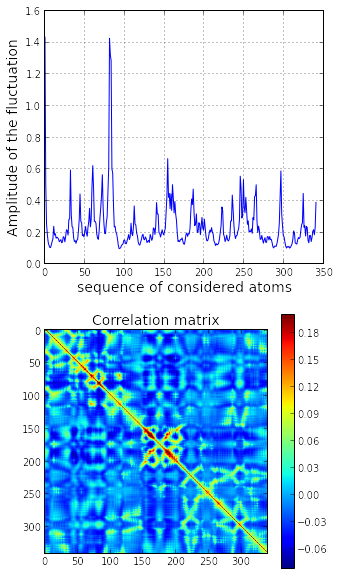
Now we can plot the first and the second mode on the structure. First I align the protein on the three axes of inertia:
R-=R.mean(axis=0)
c,a = linalg.eig(dot(R.T,R))
R = dot(R,a)And, here the plot for the first and second normal mode:
def plot_mode(w, v, mode, cte = 50,doplot=False):
samples = R
samples -= samples.mean(axis=0)
N,dim = samples.shape
if doplot:
fig = plt.figure(figsize=(10,10))
ax = fig.add_subplot(111, projection='3d')
ax.plot(samples[:,0], samples[:,1], samples[:,2], '-', markersize=10, color='green', lw=1)
ax.scatter(samples[:,0], samples[:,1], samples[:,2], c=range(samples.shape[0]), marker='o', s=64)
pc = v[:,mode].reshape(N,3)
w = sqrt(w[mode])
nstep=100
ts = linspace(-w*cte, w*cte, nstep)
interpolation=zeros((nstep,N*3))
for j,t in enumerate(ts):
interpolation[j] = samples.flatten() + t*pc.flatten()
interpolation = interpolation.reshape(nstep,N,3)
save('interpolation', interpolation)
for j,v in enumerate(pc):
x1,y1,z1 = samples[j]
x2,y2,z2 = x1+cte*w*v[0], y1+cte*w*v[1], z1+cte*w*v[2]
if doplot:
ax.plot([x1,x2], [y1,y2], [z1,z2], color='red', alpha=0.8, lw=2)
return pc
tmp = plot_mode(w, v, 6, doplot=True)
title('Normal mode #1')
tmp = plot_mode(w, v, 7, doplot=True)
title('Normal mode #2')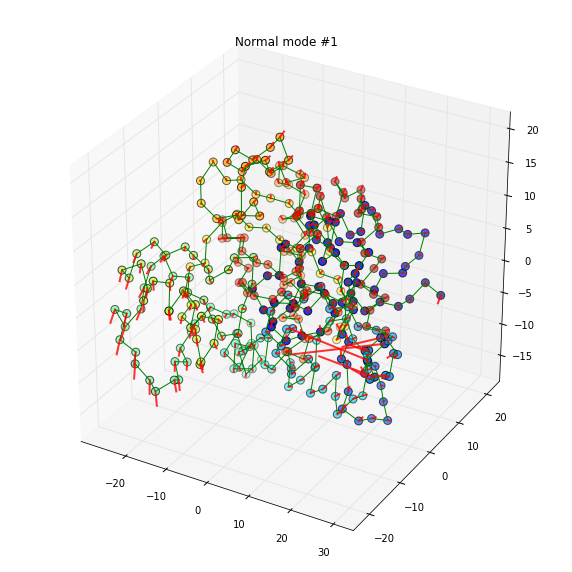
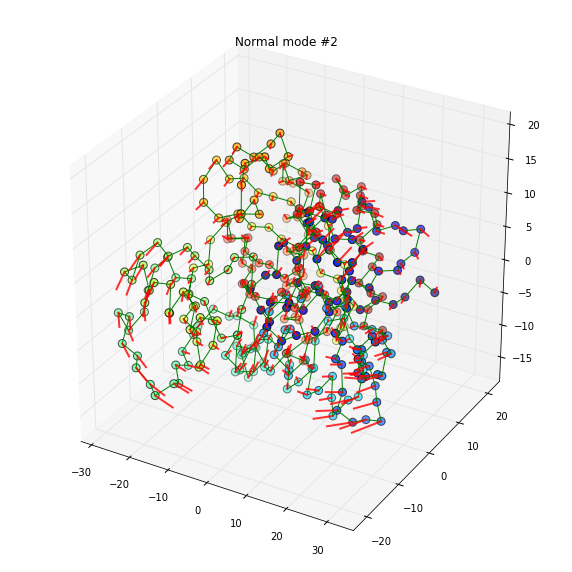
The first mode is predominated by the movement of a losely connected loop. The 2nd mode arises a more correlated global motion.
Below an interpolation along the second mode: